Ce programme calcule la justesse de prédictions de structure secondaire pour un ensemble de protéines.
![\fbox{\ttfamily
\begin{tabular}{ll}
compacc & -I[fichier prédictions] \\
& -O[fichier sortie] \\
& -V[mode verbeux]
\end{tabular} }](img204.gif)
| Options: | |
|
-I |
nom du fichier contenant les prédictions. La valeur par défaut est input.txt |
|
-O |
nom du fichier contenant le résultat. La valeur par défaut est output.txt |
|
-V |
Active le mode verbeux (sur la sortie standard ). La valeur par défaut est OFF. Pour rediriger l'information vers un fichier, utiliser compacc -V > compacc.log |
Les caractères acceptés sont H pour les résidus en
hélice-![]() , E pour les résidus en brins-
, E pour les résidus en brins-![]() et C
pour les résidus ne faisant pas partie d'une structure périodique.
et C
pour les résidus ne faisant pas partie d'une structure périodique.
Structure du fichier d'entrée (l'ordre doit être respecté):
# FIRST_CHAIN_ID SEQUENCE(1)---- $ ACTUAL_STRUCT STRUCTURE(1)--- $ METHODE1 PREDICTION(1,1) [...] $ METHODEm PREDICTION(m,1) [...] # LAST_CHAIN_ID SEQUENCE(s)---- $ ACTUAL_STRUCT STRUCTURE(s)--- $ METHODE1 PREDICTION(1,s) [...] $ METHODEm PREDICTIONm(m,s)
FIRST_CHAIN_ID SEQUENCE(1)---- ACTUAL_STRUCT STRUCTURE(1)--- METHODE1 PREDICTION(1,1) [...] METHODEm PREDICTION(m,1) [...] CONSENSUS CONSENSUS_PREDI [...]
Justesses des prédictions par chaîne:
METH1 METH2 [...] METHm CONSENSUS
CHAIN1: Q3(1,1) Q3(2,1) Q3(m,1) Q3(c,1)
CHAIN2: Q3(1,2) Q3(2,2) Q3(m,2) Q3(c,2)
[...]
CHAINs: Q3(1,s) Q3(2,s) Q3(m,s) Q3(c,s)
Chain <Q3>(1) <Q3>(2) <Q3>(m) <Q3>(c)
Stdev SD(1) SD(2) SD(m) SD(c)
Justesse globale des méthodes, toutes chaînes confondues: Pour chaque état structural:
Enfin Q3obs a la même définition qu'au-dessus mais sur l'ensemble des données. Notes Si une valeur est impossible de calculer (par exemple un coefficient de MATTHEW dans le cas où aucun résidu n'est prédit dans cet état) -99.99 est renvoyée pour cette valeur.
Le programme calcule la conservation de séquence entre séquences
homologues à chaque position d'un alignement multiple. Il calcule
d'abord les N(N-1)/2 similarités globales Sij (identités si
la matrice identité est entrée) des N séquences. Puis, pour
chaque position de l'alignement, un index de conservation est
calculé comme suit:
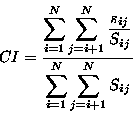
![\fbox{\ttfamily
\begin{tabular}{ll}
consindex & -A[fichier alignement] \\
& -M[matrice] \\
& -O[fichier sortie]
\end{tabular} }](img205.gif)
| Options: | |
|
-A |
Nom du fichier contenant l'alignement de séquence (défaut: alignment.txt) |
|
-M |
Nom de la matrice de comparaison, seuls les cinq premiers caractères sont évalués (défaut: default. Dans la version 1.5 default est en fait gcggap) |
|
-O |
Nom du fichier contenant le résultat (défaut: output.txt)) |
Les caractères acceptés dans les alignements sont ceux des codes non dégénérés à une lettre, en capitale.
Format de l'alignement:
C'est un format du type CLUSTAL. Chaque ligne contient l'identification de la séquence et une chaîne de résidus. La longueur totale de la ligne doit être inférieure à 100 caractères. Une ligne vide sépare les blocs de séquence.
Matrices:
Les matrices sont codées dans le fichier matrix.h, qui doit être édité pour entrer une nouvelle matrice. Tous les éléments des matrices appartiennent à l'intervalle [0-100]. Les matrices de la version 1.5 sont:
|
nucident |
matrice d'identité pour les séquences de nucléotides (ADN seulement) |
|
pepident |
matrice d'identité pour les séquences de protéines |
|
gcggap |
matrice par défaut du programme GAP du WISCONSIN package [85]. Cette matrice est une matrice de Dayhoff [79], modifiée dans Gribskov and Burgess NAR 14(16) 6745-6763 et re-échelonnée pour nos besoins. Chaque élément concernant un gap est mis à zéro. |
|
ver |
date |
notes |
|
1.0 |
09/03/1998 |
Première version |
|
1.1 |
04/04/1998 |
La sortie des similarités globales est réalisée en colonne, afin de permettre un traitement ultérieur plus facile dans des tableurs. |
|
1.2 |
17/04/1998 |
Les matrices sont maintenant dans un fichier en-tête, matrix.h, ce qui rend leur édition plus facile. |
|
1.3 |
19/05/1998 |
Erreur corrigée dans matrix.h |
|
1.4 |
29/05/1998 |
Erreurs corrigées dans matrix.h et dans le code (instructions d'utilisation). |
|
1.5 |
22/06/1998 |
Erreurs corrigées dans le choix du nom de la matrice et du fichier de sortie. |
Un article en français décrivant MELTING se trouve à l'URL: http://www.pasteur.fr/units/neubiomol/articles.html
Le programme permet de calculer l'enthalpie et l'entropie d'une hybridation entre deux acides nucléiques, et par là même de déterminer la température de fusion du duplex formé. Pour les duplex de taille inférieure à 50 nucléotides, Le calcul réalisé est physiquement ``exact'' et diffère en cela des approximations fondées par exemple sur le taux de GC.
Le programme calcule d'abord l'enthalpie et l'entropie
d'hybridation par la méthode des nearest-neighbor
à partir des paramètres élémentaires de chaque paire de CRICK
(Voir [289] pour
une revue approfondie des différents paramètres nearest-neighbor).
La différence entre les duplex commençant par A-T et G-C n'est
pas prise en compte. Quand des paramètres d'initiation différents
existent, on les moyenne. Puis la température de fusion est calculée
en utilisant la formule suivante:
Pour les sondes longues (>50nt, mais <1000nt)
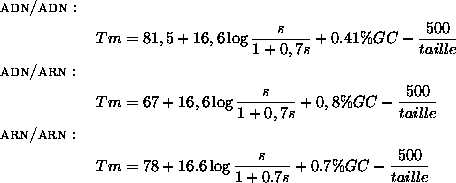
Voir [348]
![\fbox{\ttfamily
\begin{tabular}{ll}
melting & -i[fichier entrée] \\
& -o[fi...
...
& -P[concentration sonde] \\
& -N[concentration sel] \\
\end{tabular} }](img209.gif)
| Options: | |
|
-i |
Nom du fichier d'entrée. Pas de défaut |
|
-o |
Nom du fichier de sortie, par défaut melting.out. Si rien n'est spécifié mais qu'un fichier d'entrée est présent, le fichier de sortie prend le préfixe du nom du fichier d'entrée suivi de .out. |
|
-H |
Type d'hybridation, pas de défaut.
Ce paramètre doit être entré avant la séquence. Il est par exemple illégal d'entrer
la séquence sur la ligne de commande et le type d'hybridation dans un fichier d'entrée.
|
|
-S |
Pas de défaut. Attention, si la séquence dépasse 1000 nucléotides, seuls les 1000 premiers seront pris en compte. |
|
-P |
défaut: 0.0. Ce nombre peut comporter une virgule ou un point comme séparateur décimal. Attention, un nombre comme 3,525.75 sera converti en 3.525 .... La concentration en sonde doit être strictement positive et inférieure à 0,1 M. |
|
-N |
défaut: 0.0. Ce nombre peut comporter une virgule ou un point comme séparateur décimal. Attention, un nombre comme 3,525.75 sera converti en 3.525 .... La concentration en sonde doit être strictement positive et inférieure à 10 M. |
Structure d'un fichier d'entrée (l'ordre est important):
| type d'hybridation | [A,B,C,F,R,S,T,U,W] |
| séquence | [lettres capitales, longueur |
| concentration en sonde | [mol.l-1] |
| concentration en sel | [mol.l-1] |
La priorité pour les options est:
ligne de commande > fichier d'entrée > entrée interactive
Remarque: Le programme a été volontairement écrit non-permissif. Par exemple, si un paramètre illégal est entré sur la ligne de commande, mais qu'un légal est entré dans un fichier d'entrée, le programme va s'interrompre, il ne lira pas le paramètre légal.
| paire de CRICK | nombre |
| - | - |
| - | - |
| - | - |
|
ver |
date |
notes |
|
1.0 |
1997 |
Première version |
|
1.1 |
Le code a été entièrement ré-écrit. Notamment les input/output sont désormais séparés sur stdin, stdout et stderr. Cette nouvelle version prend les options sur la ligne de commande et à partir d'un fichier d'entrée. Elle produit un fichier de sortie. | |
|
2.0 |
Les paramètres pour les hybridations DNA/DNA sont mis à jour. Précedemment: Breslauer et al. (1986). Proc Natl Acad Sci USA 83 : 3746-3750. Maintenant Sugimoto et al. (1996). Nuc Acid Res 24 : 4501-4505. Pour les hybrides longs (compris entre 50 et 1000nt), un calcul approché est réalisé. | |
|
2.1 |
Les paramètres numériques peuvent avoir une virgule aussi bien qu'un point comme séparateur décimal. | |
|
2.2 |
Modularisation du code. Les paramètres calorimétriques des paires de CRICK sont maintenant inscrits dans un fichier en-tête. | |
|
3.0 |
1998 |
Il y a maintenant le choix entre plusieurs ensembles de paramètres pour les hybridations DNA/DNA. |
Le programme SSPCA étant déjà décrit de manière détaillé au chapitre 10 nous n'y reviendrons pas ici.